 今回の研究は、新エネルギー・産業技術総合開発機構(NEDO)の「次世代人工知能・ロボット中核技術開発/次世代人工知能技術分野/人間と相互理解できる次世代人工知能技術の研究開発」、科学技術振興機構(JST)の戦略的創造研究推進事業個人型研究(さきがけ)の「理論・実験・計算科学とデータ科学が連携・融合した先進的マテリアルズインフォマティクスのための基盤技術の構築」における研究課題「情報科学手法を利用した界面の構造機能相関の解明」(溝口准教授)のもとで行われたもの。
今回の研究は、新エネルギー・産業技術総合開発機構(NEDO)の「次世代人工知能・ロボット中核技術開発/次世代人工知能技術分野/人間と相互理解できる次世代人工知能技術の研究開発」、科学技術振興機構(JST)の戦略的創造研究推進事業個人型研究(さきがけ)の「理論・実験・計算科学とデータ科学が連携・融合した先進的マテリアルズインフォマティクスのための基盤技術の構築」における研究課題「情報科学手法を利用した界面の構造機能相関の解明」(溝口准教授)のもとで行われたもの。
産総研・東大生研が機械学習で物性値を高速・高精度で予測
学習結果の評価が可能な新手法、理論計算の1万倍の速度
2018.09.20−産業総合研究所機械学習研究チームの瀬々潤研究チーム長、椿真史特別研究員、および東京大学生産技術研究所物質・環境系部門の溝口照康准教授らの研究グループは19日、機械学習を利用して化学物質の分子構造から物性値を高速・高精度に予測する手法を開発したと発表した。学習に物理化学的なデータを使用することにより、ブラックボックスにならず、予測結果の妥当性の検証がしやすいことが特徴。詳細な論文は、アメリカ化学会(ACS)のThe Journal of Physical Chemistry Lettersに掲載される。
今回の研究は、新エネルギー・産業技術総合開発機構(NEDO)の「次世代人工知能・ロボット中核技術開発/次世代人工知能技術分野/人間と相互理解できる次世代人工知能技術の研究開発」、科学技術振興機構(JST)の戦略的創造研究推進事業個人型研究(さきがけ)の「理論・実験・計算科学とデータ科学が連携・融合した先進的マテリアルズインフォマティクスのための基盤技術の構築」における研究課題「情報科学手法を利用した界面の構造機能相関の解明」(溝口准教授)のもとで行われたもの。
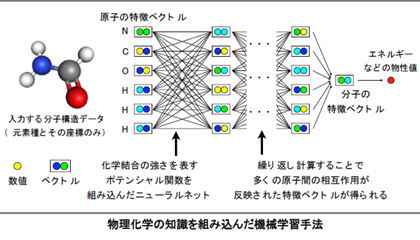
近年、分子構造からその物質の物性値を予測する機械学習の手法が発達してきているが、学習結果の解釈が難しく、その妥当性を科学的に検証しにくいという問題があった。そこで、今回の研究グループは、物理化学で用いられている近似式に基づき、分子中の原子間に働く化学結合などの相互作用の“強さの変化”を“バネの伸び縮み”であらわすポテンシャル関数を設定、それをニューラルネットで学習させた。この関数は、原子間の相互作用や化学結合の強さに対応するため、学習結果の物理化学的な解釈と検証が可能になる。
実際に、13万化合物からなるデータベースを学習させ、予測に要する計算時間と精度を評価した。その結果、例えば原子化エネルギーについては、100分の1秒で誤差0.01エレクトロンボルト以下の精度で予測できたという。精度は第一原理計算と同等で、計算時間は1万倍以上の速さだった。
また、化学結合(単結合や二重結合)の強さを示すポテンシャル曲線も、第一原理計算結果とよく一致しており、今回の手法が物理化学的に解釈できる情報をデータから学習したことを示したものだと考えられるという。研究グループでは、計算より高速に物性を評価できる利点を生かし、これまで探索されていない新物質・新材料の発見につなげていきたいとしている。
******
<関連リンク>:
産業総合研究所(人工知能研究センター機械学習研究チームのページ)
https://www.airc.aist.go.jp/mlrt/
東京大学生産技術研究所(溝口研究室のページ)
http://www.edge.iis.u-tokyo.ac.jp/